
Using Hadoop for Transcriptomics
Those of us trying to analyze next-gen sequencing data often feel constrained by the availability of computing power. Buying a very large computer (‘large’ measured by RAM size, not body mass index) is the most conventional solution, but that solution comes with a hefty price tag. Many institutions already invested heavily into distributed computing centers, and they encourage users to take full advantage of the existing resources.
Typically, distributed systems in computing clusters and supercomputing centers implement MPI-based architecture for parallel computing. Another type of distributed architecture named Hadoop/MapReduce has become popular among the internet companies processing terabytes of data. Hadoop is accessible to bioinformaticians through Amazon cloud (Elastic MapReduce), but many researchers do not understand what advantage Hadoop would provide over conventional parallel architecture. Here we explain the difference with simple examples.
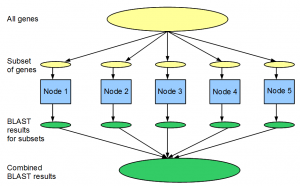
Let us say you want to run BLAST for 25,000 predicted genes against the NCBI nr protein database to determine potential homologies. If running this calculation on one computer takes 100 hours (>4 days), doing it in parallel in a cluster of 100 nodes would take only 1 hour. Most bioinformaticians are familiar with this type of parallel computation.
A more advanced application of MPI uses message passing feature of the architecture, and has been utilized by parallel genome assemblers such as ABySS.
Instead of the BLAST example mentioned above, let us think about another problem. We have a large library (100 million) of 99nt Solexa reads from a transcriptome study, and we want to find out the distribution of all 25-mers within the reads. As we explained before, de Bruijn graph approach for transcriptome assembly requires keeping tabs on all K-mers (K=25).
If we have one computer, the problem can be solved in the following manner. We go over the entire library of reads one by one, split both strands of each read into 25-mers and add the 25-mers into a giant hash table loaded into memory. After the entire library is processed, we print or save the counts for all 25-mers.
The above computation requires large amount of RAM, because we are storing all 25-mers into the memory. If our computer does not have large RAM or if we want to split the computation among many nodes, following steps can be used. (i) Split 100 million reads into 100 files with 1 million reads in each, (ii) do the counting for each file in a single node and save, (iii) sort each saved file according to 25-mers and (iii) combine 100 files into one to get distribution for all reads.
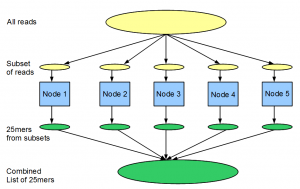
Conceptually both tasks are done by splitting the input file among 100 machines, running the computation on partitioned input file in each node and combining the results. However, there is one major difference. For BLAST, calculation on each gene is complex and CPU intensive, but it does not requires large RAM or disk-space. Moreover combining the results from 100 BLAST runs is straightforward. In case of counting 25-mers, the task on each read is very simple, but the real challenge comes from doing it on a large data set. Moreover, output from each node is a very large file. Therefore, sorting 100 such files and combining their results into one file is a major challenge. The second problem is exactly what Hadoop is designed for.
This article will not go into details of how Hadoop is implemented except on a conceptual level. Solving a hadoop problem requires two steps - (i) writing the code in Hadoop form, and (ii) finding a cluster of machine, where Hadoop architecture is implemented, and running the code.
Writing the code in Hadoop form means we have to partition the tasks into a Mapper and a Reducer function. You already know how to do it, although you did not hear the words map and reduce explicitly. In terms of the above example -
i) Map step - Partitioning the input reads into 100 subsets and divide the job of counting among 100 machines (nodes).
ii) Reduce step - Combining the sorted results from 100 nodes into one file.
If you want to use Hadoop to solve a problem, you have to come up with a way to split your large input data set among many machines, perform task in each machine and combining the results. That is all there to Hadoop.
I hope the above description helps you get started with Hadoop. In the next article of this series, we will try a hands on example and present the code for solving the problem described above.