
Finding us in homolog.us III (Search Algorithm and Indexing)
Today we shall review the indexing step of search algorithms and explain how Burrows-Wheeler transform-based algorithms (Bowtie, BWT, SOAP2) run significantly faster than alternatives like MAQ. The discussion will remain here at an abstract level, and we shall get into the details in a later commentary.
Almost all search algorithms in bioinformatics have two steps - (i) indexing of genome (reference) and (ii) searching on the indexed genome.
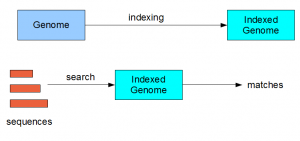
For BLAST, the indexing step is performed through ‘formatdb’ operation (improved and renamed as makeblastdb in BLAST+). Bowtie uses ‘bowtie-build’ command to build the index. Other popular programs like MAQ and BWA also have indexing steps. What does indexing do? Here we present the idea at a conceptual level.
Imagine you have a pile of books and you need to find the one on ‘Homolog.us’. You go from top to bottom of the stack and find the book that you need.
The above task is simple, if one has only ten or twelve books. Few years back, we visited the house of a retired Stanford professor in Palo Alto. Although it was not a very large place, he managed to keep 10,000 books there. Every imaginable place we looked at had boxes full of books. Even the bathroom had a large shelf with over 100 books.
With that many books, he could not have done the simple process of going through the entire pile, whenever he needed a book. People utilize two steps to speed up the search process.
i) All books are sorted in alphabetical order of titles.
See how easy it is to find ‘Homolog.us’ now.
ii) Sorting is not enough, if those 10,000 books are stored in bedroom, bathroom, kitchen, laundry room and basement. A catalog needs to be created to list -
‘A’ books are in bedroom,
‘B’ books are in living room top shelf, etc.
In a bigger pile of books, you can save two letters - ‘AA’, ‘AB’…‘TA’..and so on. This is indexing, and it can significantly speed up the search process.
Note that indexing and sorting are two different steps, and one can employ one or both to improve the search process. Even if one indexes an unsorted pile of books, that will result in significant improvement in search time than going through the entire pile of books. This is an important point, because genome sequences cannot be sorted into nucleotides, or one gets a meaningless series of ‘A’s followed by ‘C’s, ‘G’ and ‘T’s. So, the best one can do is index all regions of the genome and store locations of various K-mers. MAQ algorithm performs searches through this strategy.
Why do Burrows Wheeler transform-based algorithms run significantly faster than MAQ? Because they do allow combining of sorting and indexing together. If you remember our earlier commentary, the first column of BW transform gets the sorted genome. That, by itself, is meaningless for searches. However, when it is combined with the last column (BWT of genome), the entire sequence can be reversibly constructed.