
Dazzler Assembler for PacBio Reads - Gene Myers
you know you’re in too deep when you are watching the #AGBT14 hashtag like a day trader after an IPO - @bioinformer
We acted like those day traders, because Gene Myers, author of Celera assembler, presented on PacBio assembly in AGBT today. Thanks to tweets from @lexnederbragt, @OmicsOmicsBlog, @infoecho and many others, we got a snapshot of the talk.

Gene Myers started working on assembly problem 30 yrs ago and is returning to assembly field after 10 years. There was a big intellectual battle between Pavel Pevzner’s de Bruijn graph assembler and Myers’ OLC, and Myers conceptually combined the two approaches in his string graph paper. Obviously Myers was not happy to see de Bruijn graph stealing the show, which he described in the talk as ‘short-reads were not intellectually satisfying to me’.
-————————————————-
Edit. Corrections and Objections from Gene Myers -
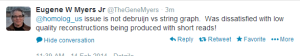
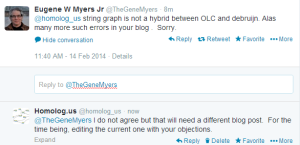
-————————————————–
The assembler he developed here is ‘PacBio-only’ and not a hybrid assembler. Possibly he does not want to touch the short reads at all, because that would require him to acknowledge that de Bruijn graphs have some value. It is better to wish those annoying short reads to go away :). For hybrid assembly, an earlier talk mentioned ECTools as very helpful.
Getting back to Myers’ talk, here are the conceptual blocks being discussed in twitter.
Conceptual blocks
1. “High error as long as its truly random is problematic w.r.t. efficiency and consensus, not quality”.
2. “sampling is perfectly Poisson and the errors are random! The location of the noise is random, Possion distribution of DNA fragments on the genome the noise does not mater “
2. 20X coverage is good enough. “Do the math. 20x coverage with 15% error will give you a Q70 base.”
3. “in some sense, string graph is answer to assembly problem”. [Notes from Homolog.us - there is no conceptual difference between de Bruijn graph and string graph. So, we hope people stop putting those two approaches as polar extremes and start to combine them.]
4. Before building string graph, he needs to take care of - “chimeric reads, contaminant reads, unclipped primer sequence, excessively erroneous reads”
5. “Everyone I know has a bigger cluster than I do.” - Dazzle’s focus is on efficiency.
Innovations
Myers solved the efficiency problem and then worked on consensus.
Here is the workflow - Overlap, scrub, correct, overlap, scrub, assemble
More detail: align at 80% -> scrub -> correct -> align at 95% -> scrub -> assemble -> consensus.
Super easy?
The main innovations are to avoid BLASR for alignment and “scrubbing” to clean up reads done using pilegrams. Details were not presented in this 10 minute talk.
Only FASTAs needed for input, quality comes in after for consensus for Quiver. Shows E.coli, Arabidopsis, Human assy times
Quiver 40 core minutes to run E.coli!
Uses fasta at beginning, at the end Quiver(-like?) polishing on raw reads
It is a low memory assembler (16GB), but it uses distributed file system.
Useful tweets:
Myers: G.bax.h5 ‘is a moose’ of a file; fasta to .dexta down to 1/14th the size.
Myers: Can correction be bypassed? A hard question. All pure strategies - PacBio only, then assemble.
Gene Myers: “If you are a bioinformatician without a distributed file system…shame on you”
needs only 16Gb of RAM but needs a distributed file system
GM, for Dazzler, No job takes more than 16G of memory, must have a distributed file system
Speed
Assembly times on non-cluster computer using dazzle: ecoli 10mins; arabidopsis 1 hr; human 5 days!!
Availability:
“Dazzler will be available on Apple app-store :)” (joke by @MeekIsaac).
Myers said the code is still not ready for public consumption and it will take couple of month to get cleaned.
Question on Transcriptome Assembly
Myers:
Q: Will you look at transcriptome assemblies?
A: A hybrid strategy would be a good thing. May not have enough PacBio copies.
Future Plan
Myers also focusing on minimizing disk consumption of pipeline #agbt14
Can do the human sapiens data set on 150Gb (would require 2Tb without compression) #AGBT14
Myers: working on compressing data from the raw data (bax.h5 file) 14 times
Myers: bypassing correction? Plugging our poster (number 211), Celera people are doing this now #agbt14
Shoutouts
Jason Chin’s previous talk & HGAP paper.